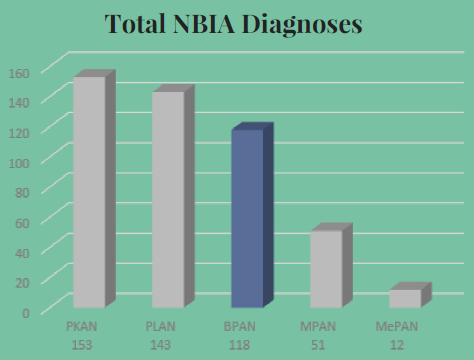
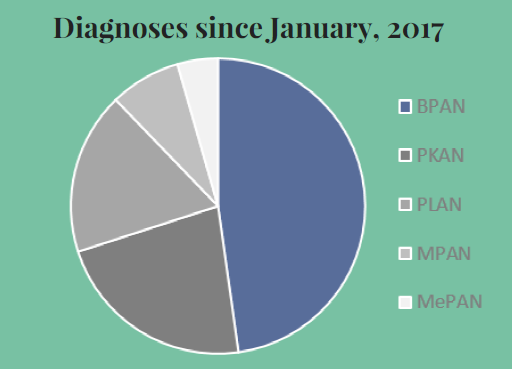
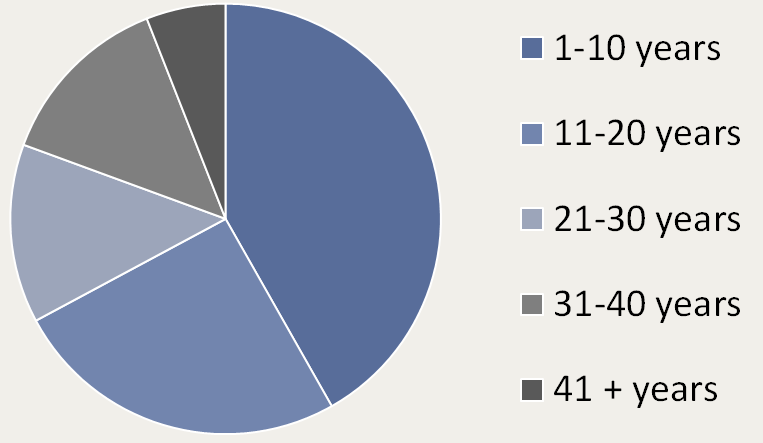
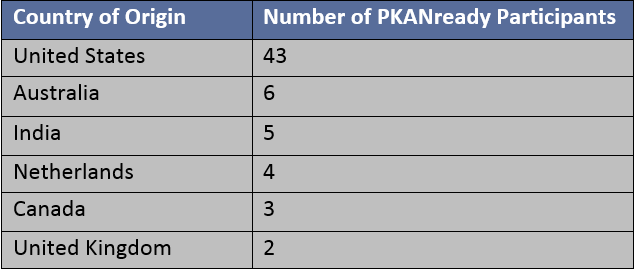
This peer-reviewed paper authored by our OHSU team in partnership with our Dutch colleagues helps explain the rationale behind our proposed CoA-Z trial in humans. In addition to a link to the paper below, we also developed some Frequently Asked Questions that summarize key points and their relevance for PKAN families.
The paper is now open for the public to read here:
4′‐Phosphopantetheine corrects CoA, iron, and dopamine metabolic defects in mammalian models of PKAN
What does the paper say?
The paper by the OHSU team presents a mouse model of PKAN. This mouse is important because it is the only one in use that has the equivalent genetic change as human PKAN patients, and is the first to show a problem with abnormal iron accumulation in brain as well as other PKAN changes. When the mice were fed a vitamin metabolite called 4’-phosphopantetheine, the biochemical abnormalities in their brains resolved. The mice did not have any dystonia; they had only biochemical changes of PKAN. But all of the biochemical changes improved after taking 4’-phosphopantetheine by mouth for two weeks. After testing the mice, Dr. Jeong and team also tested for the same changes in skin cells from people with PKAN. The same biochemical changes were found, and when those cells were bathed in 4’-phosphopantetheine, the changes resolved. The idea of trying 4’-phosphopantetheine in the PKAN mice came from work done by the group of Dr. Ody Sibon in the Netherlands.
What does this mean for people with PKAN?
The combination of positive results using 4’-phosphopantetheine in the only true mouse model of PKAN and corroborating studies in PKAN human cells suggested that 4’-phosphopantetheine might help people with PKAN. The mouse experiments showed that 4’-phosphopantetheine is not degraded in the gastrointestinal tract and that it is able to cross the blood-brain barrier. These are important discoveries that disprove concerns raised by other scientists about 4’-phosphopantetheine. The mice showed no toxic side effects from taking 4’-phosphopantetheine, which is a naturally occurring compound that is present at low quantities in most foods. The results together suggest a very low risk for toxicity from 4’-phosphopantetheine. But don’t think that you can just eat the 4’-phosphopantetheine that you need to help your PKAN. The levels in food are so low that an adult would need to eat 100 lbs of meat every day in order to get enough 4’-phosphopantetheine.
Is 4’-phosphopantetheine the same as CoA-Z?
CoA-Z contains 4’-phosphopantetheine as one of its ingredients. The formulated compound is called CoA-Z, and the active ingredient is 4’-phosphopantetheine. CoA-Z contains other inactive ingredients as well.
Where can I get 4’-phosphopantetheine?
As far as we know, 4’-phosphopantetheine that is suitable for human use is not available anywhere for purchase; you should be wary of any product making such claims. Any 4’-phosphopantetheine that might be available to buy has not been synthesized in a way that makes it safe for human use, and it is very expensive. The 4’-phosphopantetheine that we synthesized for use in the human clinical trial is ultra-pure and was made following strict safety procedures. The synthesis of this special 4’-phosphopantetheine was funded by the Spoonbill Foundation, Stichting Lepelaar, and “ZZF”, the Dutch Foundation for rare diseases, using funds donated by families and other supporters. This high grade 4’-phosphopantetheine is only available to participants in the clinical trial and is not available for purchase.
When will I be able to buy 4’-phosphopantetheine?
The clinical trials must be completed and approval given by FDA before 4’-phosphopantetheine can be made available for purchase.
When can you tell us more about the human clinical trial of CoA-Z?
We will provide detailed information to the PKAN family community starting in the coming weeks. For families outside of the US and Canada, we will be providing updates on trials in other countries as progress is made and information becomes available. We know how eager you all are!