If you or a loved one has recently been diagnosed with MEPAN, we hope our website can help answer some of your questions and guide you to the most appropriate resources. We encourage you to call our team to speak to a genetic counselor who can help you navigate this diagnosis. Please email us at info@nbiacure.org and we will set up a phone call with you.
MEPAN (Mitochondrial Enoyl CoA Reductase Protein-Associated Neurodegeneration) is a rare disorder that is characterized by a progressive childhood-onset movement disorder and optic atrophy. Although we now know MEPAN does not appear to involve brain iron accumulation, both the symptoms and the brain regions involved overlap with NBIA. For these reasons, our team was involved in the gene discovery and continues to have an interest in this NBIA “mimic.”
SYMPTOMS
The movement disorder symptoms of MEPAN usually start to appear during childhood (before age 7). Decreased vision typically begins in childhood and can lead to blindness in adulthood.
Because there are currently only 13 individuals known to have MEPAN, the full spectrum of symptoms and progression have not been defined yet.
Common symptoms include:
Dysarthria (poor articulation or slurring of speech)
- Progressive speech problems
- Can make it difficult to be understood by acquaintances and others in the community. Usually people close to the individuals with MEPAN have learned to better understand their speech
Involuntary movements
- Most common symptom is dystonia (involuntary muscle contraction and spasms)
- Chorea and/or ataxia may also occur
- Typically start to develop between 15 months – 6.5 years of age
- Over time some individuals require a walker or wheelchair for ambulation; others may never learn to walk
Eye findings
- Optic atrophy (deterioration of the nerve that connects the eye to the brain)
- Manifest as reduced visual acuity in childhood
- Can lead to functional blindness in adulthood in some individuals
- Nystagmus (roving eye movements) can also be seen
Cognition
- Intellect is often (but not always) preserved
ETHNICITY
MEPAN is more frequent in individuals of Ashkenazi Jewish ancestry. However, it has also been described in individuals of other ethnicities.
CAUSE/GENETICS
 The human body is made up of millions of cells. Inside every cell there is a structure called DNA, which is like an instruction book. DNA contains detailed steps about how all the parts of the body are put together and how they work. However, DNA contains too much information to fit into a single “book” so it is packaged into multiple volumes called chromosomes. Humans typically have 46 total chromosomes that are organized in 23 pairs. There are two copies of each chromosome because we receive one set of 23 chromosomes from our biological mother and the other set of 23 from our biological father. Chromosomes 1-22 are called autosomes and the last pair is called the sex chromosomes because they determine a person’s gender. Females have two X chromosomes and males have one X and one Y.
The human body is made up of millions of cells. Inside every cell there is a structure called DNA, which is like an instruction book. DNA contains detailed steps about how all the parts of the body are put together and how they work. However, DNA contains too much information to fit into a single “book” so it is packaged into multiple volumes called chromosomes. Humans typically have 46 total chromosomes that are organized in 23 pairs. There are two copies of each chromosome because we receive one set of 23 chromosomes from our biological mother and the other set of 23 from our biological father. Chromosomes 1-22 are called autosomes and the last pair is called the sex chromosomes because they determine a person’s gender. Females have two X chromosomes and males have one X and one Y.
If DNA is the body’s instruction book and it is stored in multiple volumes (called chromosomes), then genes would be the individual chapters of those books. Genes are small pieces of DNA that regulate certain parts or functions of the body. Sometimes multiple genes (or chapters) are needed to control one function. Other times, just one gene (or chapter) can influence multiple functions. Since there are two copies of each chromosome, there are also two copies of each gene. In some gene pairs, both copies need to be expressed (or turned on) in order for them to do their job correctly. For other genes pairs, only one copy needs to be expressed.
When a single cell in the human body divides and replicates, its DNA is also replicated. This replication process is usually very accurate but sometimes the body can make a mistake and create a “typo” (or mutation). Just like a typo in a book, a mutation in the DNA can be unnoticeable, harmless, or serious. A mutation with serious consequences can result in a part of the body not developing correctly or a particular function not working properly.
In the case of NBIA disorders, changes in certain genes cause a person to develop their particular type of NBIA. Changes in these NBIA genes lead to the groups of symptoms we observe, although we do not yet understand how the changed genes cause many of these findings. MECR is the only gene known to cause MEPAN. MECR’s main job is to tell the body’s cells how to make a protein that completes the last step in mitochondrial fatty acid synthesis, which turns trans-2-enoyl-ACP into acyl-ACP. MECR also serves as the precursor for lipoic acid synthesis which functions as a cofactor for key enzymes of the respiratory chain. Therefore, decreased MECR activity reduces mitochondrial RNA processing and translation, as well as respiratory complex assembly.
As mentioned earlier, humans have a total of 23 pairs of chromosomes. Half of these chromosomes are passed down (or inherited) from the biological mother and half from the biological father. The way in which a gene carrying a change is passed down from parents to child varies from gene to gene. The MECR gene that is altered in those with MEPAN is inherited in an autosomal recessive manner.
“Autosomal” refers to the fact that the MECR gene is located on chromosome 1, which is one of the autosomes (chromosome pairs 1-22). Since the sex chromosomes are not involved, males and females are equally likely to inherit the changed gene. “Recessive” refers to the fact that a gene change must be present in both copies of the MECR gene for a person to have MEPAN. If an individual has only one MECR gene change, then they are called a “carrier” for MEPAN. Carriers do not have health problems related to that gene change and often do not know they carry a recessive gene change. However, if two MEPAN carriers have a child together, then there is a 25% chance that they will both pass on their recessive MECR gene changes and have a child with MEPAN.
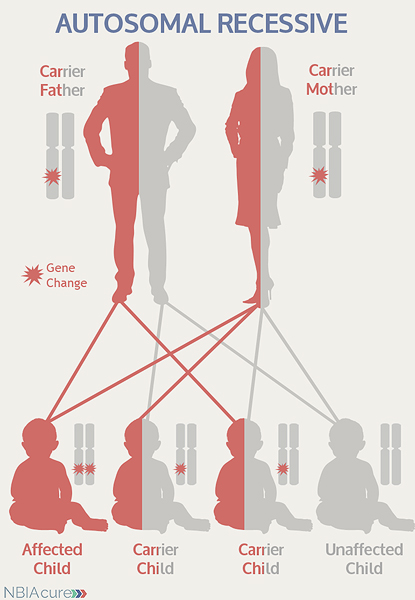 As seen in the image to the left, in a pregnancy between two MEPAN carriers:
As seen in the image to the left, in a pregnancy between two MEPAN carriers:
- There is a 25% chance of the child having MEPAN
- There is a 50% chance that the child will be a carrier like his/her parents
- There is a 25% chance that the child will not have MEPAN or be a carrier
DIAGNOSIS & TESTING
Diagnosis of MEPAN is confirmed through genetic testing of the MECR gene to find two gene changes.
If no gene change or only one gene change is found through sequence analysis of the MECR gene, then deletion/duplication analysis could be considered. However, to date no exonic or whole gene deletions have been reported.
Very rarely, an individual with the signs and symptoms of MEPAN will have only one or even no MECR gene changes identified. This can happen because genetic testing is not perfect and has certain limitations. It does not mean the person does not have MEPAN; it may just mean we do not yet have the technology to find the gene change(s). In these cases it becomes very important to have doctors experienced with MEPAN review the MRI and the person’s symptoms very carefully. Since MEPAN is still a relatively new and rare diagnosis, it is likely that testing may change over time and that the symptoms of MEPAN will become better understood and potentially more recognizable in the coming years.
MRI FINDINGS
A brain MRI is a standard diagnostic tool for all NBIA disorders. MRI stands for magnetic resonance imaging. An MRI produces a picture of the body that is created using a magnetic field and a computer. The technology used in an MRI is different from that of an x-ray. An MRI is painless and is even considered safe to do during pregnancy. Sometimes an MRI is done of the whole body, but more often, a doctor will order an MRI of one particular part of the body
MRI findings for MEPAN include:
- Bilateral hyper-intense T2 signal in one or more structures of the basal ganglia (i.e., caudate, putamen or pallidum) evident around the onset of dystonia
MANAGEMENT
 There is no standard treatment for MEPAN. Patients are managed by a team of medical professionals that recommends treatments based on current symptoms.
There is no standard treatment for MEPAN. Patients are managed by a team of medical professionals that recommends treatments based on current symptoms.
After diagnosis, individuals with MEPAN are recommended to get the following evaluations to determine the extent of their disease:
- Ophthalmology evaluation to assess for optic atrophy and visual acuity
- Neurological examination for dystonia
- Neuropsychological examination to assess cognitive function
- Assessment for physical therapy, occupational therapy, and/or speech therapy
- Medical genetics consultation
Therapies and medications that can be considered:
- Visual aids can be used in cases of decreased visual acuity
- Physiotherapy can be used to maintain range of movement
- Special aids such as braces, walkers and wheelchairs can maintain/improve mobility
- Speech therapy, if speech dysarthria is present, and assessment for augmentative communication devices
- Medications which may relieve dystonia, such as anticholinergic agents, baclofen and benzodiazepines, can be considered
- Anticholinergic agents act peripherally on the on the neuromuscular junction, but can have a variety of adverse central nervous system (CNS) effects
- Baclofen, which works on GABAB receptors and functions as a CNS depressant and skeletal muscle relaxant, can be administered either through a spinal pump or systemically
- Benzodiazepines, which are GABAA agonists, can reduce muscle tone and alleviate the dystonia; however, they also cause sedation
- While some patients with severe dystonia are treated with DBS, experience with MEPAN is limited and, to date, there are no reports of individuals with this disorder treated with DBS. Moreover, due to the existence of basal ganglia lesions, DBS may not be suitable for many individuals with MEPAN
Long-term management for MEPAN should include:
- Yearly eye exams
- Yearly neurological assessment to determine need for additional interventions, including speech therapy
PROGRESSION
It is unknown what kind of impact MEPAN has on life expectancy. All 10 of the individuals currently known to have this condition are still alive and two of the oldest are in their 40s and 50s.
Copyright © 2014 by NBIAcure.org. All rights reserved.