The PKAN physician-scientists at OHSU and scientists from the Horae Gene Therapy Center at UMass Chan Medical school have recently formed a partnership to develop gene therapy for PKAN. Early work in PKAN mice was initially done by Dr. Lauriel Earley, a former graduate student in Molecular & Medical Genetics at OHSU. Dr. Earley’s work was completed in the Hayflick lab after she departed to start a new position in industry. Now, using the preliminary mouse studies as a launching pad, OHSU and UMass are working to move the project forward. These experts from OHSU and UMass bring complementary skill sets, decades of experience, and a collaborative approach to the partnership. Meanwhile, the Loving Loic Foundation is working tirelessly to raise the necessary funds for the project. The Loving Loic Foundation was established in late 2023 by the Blackford family in honor of their son, Loic, who had recently been diagnosed with PKAN. Their mission is to fund research for developing a lifetime cure for PKAN for all children through the development of an effective gene therapy. The Loving Loic Foundation works closely with the NBIA Disorders Association and other advocacy groups. Donations to support this project can also be made to a dedicated PKAN Gene Therapy Research Fund through the OHSU Foundation.
All posts by Puneet Rai
CoA-Z Trial Update
With the new year, we’d like to send an update on our progress with CoA-Z. In September, we told you that we had decided to set aside plans for further clinical trials with CoA-Z, instead focusing our efforts on compiling the necessary data to present to the FDA in order to gain approval to make the compound more widely available as quickly as possible. This was based on our belief that our data was strong enough to submit without doing any additional studies. We still very much believe this to be true.
The FDA will be looking at the data from the clinical trial to decide whether CoA-Z is safe and whether it changes a biological marker of PKAN in the blood. As you know from our previous communications, the trial data showed that CoA-Z did change the blood biomarker just as we had hoped, and it did it in a ‘dose-dependent’ way, meaning that higher doses produced a larger response.
To demonstrate safety, the FDA requires that we show in an objective way that there is no difference between the safety of the compound being tested (i.e. CoA-Z) compared with placebo, and between one dose compared with another: this is why the first 6 months of the trial had to be done in a ‘double-blind, placebo-controlled’ way (meaning nobody knew who was taking a placebo and who was taking CoA-Z). Now that we’ve finished the analysis of data from this part of the study we are confident that CoA-Z was safe and well-tolerated at all doses tested. We are finalizing the report from this phase of the study and preparing to submit it to the FDA, along with a lot of other required information about things such as the CoA-Z manufacturing process, its stability when stored in different conditions, its effects in mice, and much, much more. Our goal is to submit a draft of our application to the FDA by the end of March.
While it is impossible to predict how long the FDA will take to complete their review of the draft, they have indicated that this ‘pre-review’ before final submission may reduce the risk of delays later on, and so we are grateful for the agency’s time and attention to our submission. In the interim, we will shift our focus to analyzing data from the second segment of the trial, called the ‘open-label’ portion (when everyone was taking the same dose of CoA-Z) so this is ready to include in the final submission. For those of you who were in the trial, it is only natural to wonder which group you or your child was in during those first 6 months. After we get our final application in to the FDA, we will have access to this information ourselves and will contact everyone privately to share these details. For now, please do not contact us about this—we will reach out once we have the work completed and submitted.
We are very hopeful our data will convince the FDA that no additional studies will be needed. We remain very grateful to the PKAN community for all of your support and encouragement.
Update to the PKAN Community on CoA-Z
At the NBIA Family Meeting in May and in our follow-up communication to CoA-Z study participants, we mentioned the possibility of our doing one or more short follow-up studies of CoA-Z to supplement our clinical trial data. After further data analysis and reflection, we have decided that our current data is strong enough to submit to the FDA without doing any additional clinical studies. If the FDA agrees, then this will be the fastest path to receiving the approvals we need to make the compound more widely available to the PKAN community.
So we are setting aside any plans for follow-up studies for now, instead directing all our efforts to compiling our data to present to FDA later this fall. It is a big task, because it not only involves data from the CoA-Z clinical trial, but also from all the related background work going back to 2015: more than 500 pages in length! We don’t have the resources to offer further access to CoA-Z before FDA approval, and so we feel this is the best path to make it broadly available to the PKAN community.
The studies we were considering would have posed quite a burden on participants (such as several blood draws over a short time period) and the studies would not have been long enough to offer any real expectation of benefit, so we hope the PKAN community will see this as good news – we do! We know the community is deeply interested in our progress, and we will continue to update you.
– The OHSU CoA-Z trial team
Statement about Rett syndrome medication
A new medication for Rett syndrome, called Daybue, was recently approved by the FDA. Since there are some clinical similarities between BPAN and Rett syndrome, including regression, seizures, intellectual disability, motor changes and certain repetitive behaviors, it is natural to wonder whether Daybue could help people with BPAN. We at OHSU continue to ask ourselves this question and are reviewing information and data that may help inform our thinking about this. It is important to understand that the common Rett gene, MECP2, and the medication in Daybue have a direct link and rationale that explain why Daybue works. This specifically relates to a protein called insulin-like growth factor 1 (IGF1) that is low in Rett syndrome and restored by Daybue. In BPAN, however, there is currently no evidence that IGF1 is reduced or plays a role in disease development. Still, it is worth exploring whether Daybue could benefit the BPAN population, starting with studies in BPAN animal or cell-based models. Even if it cannot, the lessons learned from the approaches to treating Rett syndrome, which is also X-linked, will likely be useful to BPAN investigators. For these reasons, we stay informed about Rett syndrome studies with an eye towards applications for BPAN.
CoA-Z Clinical Trial Update
It’s hard to believe it, but the CoA-Z clinical trial started way back in December 2019 when we had no idea how much all our lives were about to be upended by a global pandemic. Many of you have reached out since then to ask for updates and to find out when results will be released. As we approach the two year anniversary, we thought this would be a good opportunity to update the community on how things have been going.
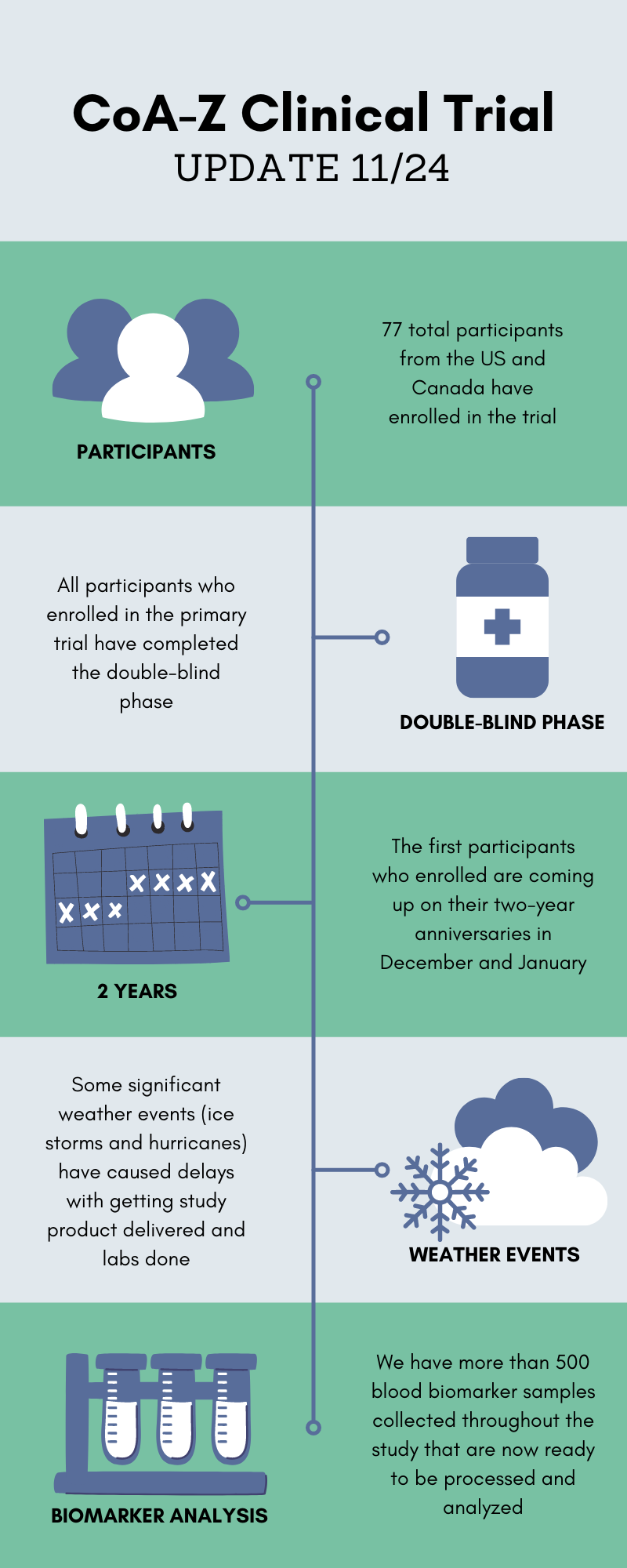
You may recall that the trial was originally planned to be two years in duration for each participant. Since the double-blind phase is now completed and many participants are approaching their key two year study visit, our team has started making plans for data analysis. We know you are all anxious to see the results, but we want to prepare you that this process will take some time. The blood biomarker analysis is one of the most important parts of the study and we have hundreds of samples that must first be processed with great care and consistency.
Overall our team is pleased with how the trial has run for the past two years. We are very grateful for everyone’s commitment to get their study visits done despite their busy lives, unexpected weather events, and even a pandemic. We will share more updates as soon as we can!
BPAN Management Guideline and Podcast
A consensus management guideline for BPAN has been published in Developmental Medicine & Child Neurology that will help support clinicians by guiding the diagnostic process, management and surveillance at different ages and stages of BPAN. We hope it will serve as a valuable resource for families who often become advocates for their children.
Update on BPAN
A scientific paper about BPAN recently published in the journal Autophagy has created some confusion within the community of BPAN families, friends, and caregivers. Entitled “WDR45, one gene associated with multiple developmental disorders,” the paper suggests that gene mutations in WDR45 lead to six separate disorders rather than just BPAN. As you can imagine, this suggestion could make parents question whether their child has the right diagnosis. This paper was written by a team that reviewed the available literature, but has limited clinical experience working with BPAN.
As the team that discovered the gene for BPAN and has deep clinical experience working with patients and running the largest natural history study, we want to reaffirm our strong belief that BPAN is the only disorder associated with gene mutations in WDR45. As you know, BPAN is complicated by several factors:
All these factors lead to a very wide range of variation in BPAN. We see variation in age of onset, motor skills, intellectual function, language, presence or absence of seizures, seizure types, and more. However, equally important, we also see overlap among individuals with BPAN that link them all to their common gene. While a child with BPAN may have various descriptions used, such as “Rett-like” or “epileptic encephalopathy,” their primary diagnosis and the underlying cause of their disease is still BPAN.
Unfortunately, this paper also has a significant error in describing a key MRI finding in BPAN. We sincerely hope the authors will take steps to rectify this with the journal so the community of readers can be notified.
BPANready Update & Feb Newsletter
This article and other news can be found in the latest issue of our newsletter which can be downloaded/viewed here: February 2021.
In May, BPANready turned 2 years old! It has been the fastest enrolling of our natural history studies. We broke 100 participants in last February 2020! We have also received 69 research blood samples and there have been nearly 400 study visits. None of this would have been possible without the BPAN family community’s enthusiastic research participation, and the grant funding we received from the NBIA Disorders Association and the University of Pennsylvania’s Orphan Disease Center Million Dollar Bike Ride. While the number of participants is important for the strength of a natural history study, so too is the number of years of data for each participant. We encourage everyone enrolled to keep inputting their data! We know that it can seem repetitive, but that is exactly what we need so we can learn how BPAN changes over time. Only with that background knowledge will we be able to tell if future therapeutics are changing the course of the disease. We need to be “BPAN ready” for treatment trials! We are currently exploring how to fund the BPANready study going forward. Our preliminary data analysis from the first two years has told us we are on the right track with our approach, but we need to continue to collect your family member’s data and samples over time.
The not-so-good news is that we are facing a funding crisis for BPANready. While we have some funding support for the PKANready and PLANready natural history studies, without further funding for BPANready, we will need to curtail our efforts on the study. This would have a detrimental impact on this important work, and while we are working hard to sustain this work, we need the family community’s support and funding. We have been exploring how to fund the BPANready study going forward, and will continue to do so. If families want to donate, they can do so here: Click HERE to Donate.
COVID-19 Vaccination for kids and adults with NBIA
Many families have asked us recently whether their children ages 16+ with NBIA should be vaccinated for COVID-19. Since the available vaccines have not yet been approved for kids under 16, it will not yet be a question for many of you. However, others with NBIA have already been offered vaccination or will be in the near future.
The NBIAcure group has always supported vaccination for those with NBIA, including a regular pediatric vaccination schedule and annual flu shots. While vaccination has never been studied specifically in NBIA, the information we have available and our decades of experience with these disorders suggest that the benefit of COVID-19 vaccination will far outweigh potential risks. Some parents have specifically voiced concerns about their children who have seizures. If your loved one with NBIA has seizures or an increased risk for seizures, then mindfulness around potential fever is warranted. Mild fever, as well as headache and general achiness, is more likely after the second dose of vaccine, but can also happen after the first. These symptoms are an expected reaction to the vaccine, and are considered a good sign that the immune system is responding normally. Recommendations regarding pre-medication with a fever-reducing medication like acetaminophen (Tylenol, paracetamol) vary, but because of increased risk of seizures with fever, we encourage you to discuss this with your loved one’s healthcare provider. All the vaccines approved for use at the time of writing are a two-dose series. Although side effects are more common after the second dose, it is very important to follow through with the second dose as your loved one will not be fully protected without it.
Our team has enthusiastically welcomed the advent of vaccination and feels it’s a huge step forward in our journey of the past 10 months. As a side note, the fact that the vaccines are based on basic tenets of genetics has been particularly exciting! While they are new, mRNA vaccines promise to be highly effective and will likely continue to be used in the future. They leverage the machinery our cells already have to read genetic messages and build proteins. In the case of the vaccine, a new instruction or “recipe” is provided and our cells are able to do the rest. These vaccines do not change the natural genetic material already within our cells and they do not cause COVID-19 infection. We hope this message offers the community some hope and reassurance. We look forward to being able to see you in person in the future.
A paper from the Sibon lab reveals important insights into biochemical changes in PKAN and related disorders
Dr. Sibon’s group in the Netherlands has published a paper that reveals important insights into the biochemical changes in PKAN and related disorders (CoPAN, MePAN and PDH-E2 deficiency). It reports work done largely by Roald Lambrechts (who just got his PhD yesterday!) and others in the Sibon lab. Dr. Sibon has also written a FAQs section explaining what the paper means.
The paper is now open for the public to read here:
CoA‐dependent activation of mitochondrial acyl carrier protein links four neurodegenerative diseases
What does the paper say?
In order to develop a treatment for a disease, it is important to understand the symptoms of a disease. The manuscript by Lambrechts et al explains some of the characteristics of PKAN and explains why some symptoms of PKAN are comparable to symptoms of 3 other diseases. These other diseases are CoPAN, MePAN and PDH-E2 deficiency. The presence of similar symptoms suggests that the 4 diseases share comparable metabolic defects. The manuscript describes and explains these shared metabolic defects in a fruitfly model and in human cells. One of the key findings of the manuscript is that the protein mtACP is less active in PKAN. A defect in mtACP has never been considered nor investigated in relation to PKAN and this provides important insights why mutations in PANK2 gene cause the disease PKAN. A less active mtACP explains also the metabolic defects observed in cells of PKAN patients and the iron accumulation reported in the manuscript of the OHSU group in the PKAN mouse model. The results presented in the Lambrechts et al manuscript also explain why 4-phosphopantetheine corrects the metabolic defects in PKAN as elegantly demonstrated in the Jeong et al manuscript.
What does it mean for people with PKAN?
The manuscript further supports the findings of the OHSU group that the mouse model is a valuable model to understand PKAN and is in agreement with the findings of the OHSU group that 4-phosphopantetheine might help people with PKAN.
What does it mean for people with CoPAN, MePAN and PDH-deficiencies?
Based on the results of the fly model, the results suggest that 4-phosphopantetheine most likely is not promising for CoPAN, MePAN and PDH-E2 deficiency patients. For these diseases the manuscript suggests other treatment strategies. These strategies need to be further explored before they can be implemented in the clinic.