If you or a loved one has recently been diagnosed with PLAN/INAD, we hope our website can help answer some of your questions and guide you to the most appropriate resources. We encourage you to call our team to speak to a genetic counselor who can help you navigate this diagnosis. Please email us at info@nbiacure.org and we will set up a phone call with you.
PLAN (PLA2G6-associated neurodegeneration) is a NBIA (neurodegeneration with brain iron accumulation) disorder. Based on an individual’s age of onset and symptoms, they may be classified as having one of three types of PLAN: INAD, aNAD or PLA2G6-related dystonia-parkinsonism. However, as we learn more about PLAN, we have found that some people fall between these 3 categories. In other words, there is a broad spectrum of symptoms.
SYMPTOMS
The symptoms of INAD (infantile neuroaxonal dystrophy) usually start to appear between the ages of 6 months and 1 year. A common pattern in young children is loss of skills and progression of the disorder over time.
Common symptoms include:
Psychomotor (mental and physical ability) regression or delay
- Loss of previously acquired milestones
- Delayed walking or gait disturbance (difficulty with walking)
- Typically the first symptom that is noticed
Ataxia (loss of muscle control)
- Typically one of the first symptoms that is noticed
Truncal hypotonia (low muscle tone in the trunk more than the limbs)
- Occurs early in the disease
- Followed by later development of spastic tetraparesis (muscle tightness and weakness in both arms and legs)
- Hypperflexia (overactive reflexes) seen at the beginning of the disease
- Areflexia (absent reflexes) seen later in the disease
Eye features
- Strabismus (crossed eyes) & nystagmus (rapid uncontrolled eye movements)
- Seen during early childhood
- Optic atrophy (deterioration of the nerve that connects the eye to the brain)
- Seen later in the disease
Bulbar dysfunction (impairment of the function of cranial nerves IX, X, XI and XII)
- Results in speech problems
- Dysarthria (poor articulation or slurring)
- Dysphonia (defective use of the voice)
- Dysphasia (difficulty in using or understanding words)
- Causes nutritional problems
- Dysphagia (difficulty swallowing)
- Difficulty with chewing
- Choking on liquids
- Nasal regurgitation
Seizures
- Occur in some individuals
- Develop later in the disease
Progressive cognitive (mental) decline
- Seen during the later stages of disease
CAUSE/GENETICS
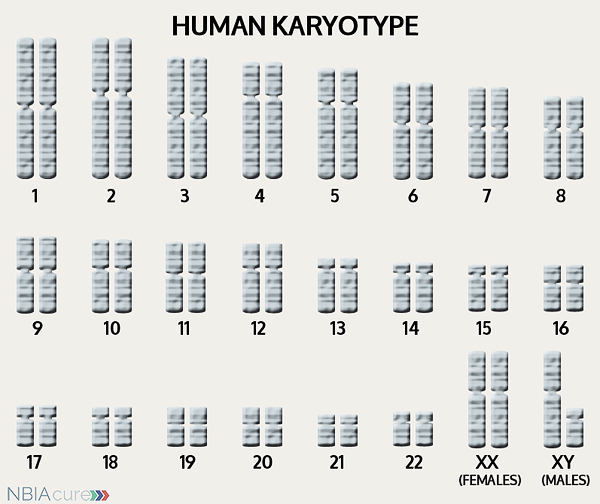 The human body is made up of millions of cells. Inside every cell there is a structure called DNA, which is like an instruction book. DNA contains detailed steps about how all the parts of the body are put together and how they work. However, DNA contains too much information to fit into a single “book,” so it is packaged into multiple volumes called chromosomes. Humans typically have 46 total chromosomes that are organized in 23 pairs. There are two copies of each chromosome because we receive one set of 23 chromosomes from our biological mother and the other set of 23 from our biological father. Chromosomes 1-22 are called autosomes and the last pair is called the sex chromosomes because they determine a person’s gender. Females have two X chromosomes and males have one X and one Y.
The human body is made up of millions of cells. Inside every cell there is a structure called DNA, which is like an instruction book. DNA contains detailed steps about how all the parts of the body are put together and how they work. However, DNA contains too much information to fit into a single “book,” so it is packaged into multiple volumes called chromosomes. Humans typically have 46 total chromosomes that are organized in 23 pairs. There are two copies of each chromosome because we receive one set of 23 chromosomes from our biological mother and the other set of 23 from our biological father. Chromosomes 1-22 are called autosomes and the last pair is called the sex chromosomes because they determine a person’s gender. Females have two X chromosomes and males have one X and one Y.
If DNA is the body’s instruction book and it is stored in multiple volumes (called chromosomes), then genes would be the individual chapters of those books. Genes are small pieces of DNA that regulate certain parts or functions of the body. Sometimes multiple genes (or chapters) are needed to control one function. Other times, just one gene (or chapter) can influence multiple functions. Since there are two copies of each chromosome, there are also two copies of each gene. In some gene pairs, both copies need to be expressed (or turned on) in order for them to do their job correctly. For other genes pairs, only one copy needs to be expressed.
When a single cell in the human body divides and replicates, its DNA is also replicated. This replication process is usually very accurate but sometimes the body can make a mistake and create a “typo” (or mutation). Just like a typo in a book, a mutation in the DNA can be unnoticeable, harmless, or serious. A mutation with serious consequences can result in a part of the body not developing correctly or a particular function not working properly.
In the case of NBIA disorders, changes in certain genes cause a person to develop their particular type of NBIA. Changes in these NBIA genes lead to the groups of symptoms we observe, although we do not yet understand how the changed genes cause many of these findings. PLA2G6 is the only gene known to cause all the types of PLAN. PLA2G6’s main job is to tell the body’s cells how to make an enzyme called A2 phospholipase that breaks down phospholipids. When a change the PLA2G6 gene impairs the function of this enzyme, the cells’ membrane maintenance is disrupted and may lead to the development of spheroid bodies in the nerve axons. It is not yet clear to us how the decrease in A2 phospholipase eventually leads to iron accumulation in the brain.
As mentioned earlier, humans have a total of 23 pairs of chromosomes. Half of these chromosomes are passed down (or inherited) from the biological mother and half from the biological father. The way in which a gene carrying a change is passed down from parents to child varies from gene to gene. The PLA2G6 gene that is altered in those with INAD is inherited in an autosomal recessive manner.
“Autosomal” refers to the fact that the PLA2G6 gene is located on chromosome 22, which is one of the autosomes (chromosome pairs 1-22). Since the sex chromosomes are not involved, males and females are equally likely to inherit the changed gene. “Recessive” refers to the fact that a gene change must be present in both copies of the PLA2G6 gene for a person to have INAD. If an individual has only one PLA2G6 gene change, then they are called a “carrier” for INAD. Carriers do not have health problems related to that gene change and often do not know they carry a recessive gene change. However, if two INAD carriers have a child together, then there is a 25% chance that they will both pass on their recessive PLA2G6 gene changes and have a child with INAD.
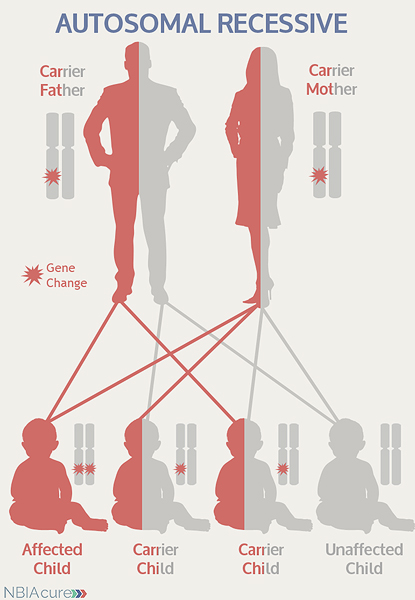 As seen in the image to the left, in a pregnancy between two INAD carriers:
As seen in the image to the left, in a pregnancy between two INAD carriers:
- There is a 25% chance of the child having INAD
- There is a 50% chance that the child will be a carrier like his/her parents
- There is a 25% chance that the child will not have INAD or be a carrier
DIAGNOSIS & TESTING
An MRI of the brain and an ophthalmologic exam are key tests used to establish the clinical symptoms of INAD. MRI stands for magnetic resonance imaging. An MRI produces a picture of the body that is created using a magnetic field and a computer. The technology used in an MRI is different from that of an x-ray. MRI is painless and is even considered safe to do during pregnancy. Sometimes an MRI is done of the whole body, but more often, a doctor will order an MRI of one particular part of the body.
An MRI is often ordered as part of the work-up of children with INAD symptoms. However, it may be normal, especially early in disease. A T2 sequence is the preferred type of MRI because it is highly sensitive to the detection of brain iron.
MRI findings for INAD include:
- Cerebellar changes
- Cerebellar atrophy (degeneration of the cerebellum)
- Cerebellar hyperintensities (brightness)
- Hypointensity (darkness) in globus pallidus
- Indicates iron accumulation
- Not all children with INAD have iron accumulation
- White matter abnormalities
Before genetic testing was available, other tests were used to try to confirm a diagnosis of INAD:
- Biopsy of one or more of the following tissues to look for axonal spheroids in the nerves:
- Conjunctiva (outer layer of the eye)
- Skin
- Rectum
- Muscle
- Other peripheral nerve
Now, a diagnosis of INAD is confirmed through genetic testing of the PLA2G6 gene to find two gene changes. At least one PLA2G6 gene change is found through DNA sequence analysis in ~85% of individuals.
If no gene change or only one gene change is found through sequence analysis of the PLA2G6 gene, then genetic testing may proceed to deletion/duplication analysis. Among individuals who go forward with deletion/duplication analysis, a gene change may be found in ~12.5% of cases.
Sometimes an individual with the signs and symptoms of INAD will have only one or even no PLA2G6 gene changes identified. This can happen because the genetic testing is not perfect and has certain limitations. It does not mean the person does not have INAD; it may just mean we do not yet have the technology to find the hidden gene change. In these cases it becomes very important to have doctors experienced with INAD review the MRI and the person’s symptoms very carefully to be as sure as possible of the diagnosis.
MANAGEMENT
 There is no standard treatment for INAD. Diagnosed individuals are managed by a team of medical professionals that recommends treatments based on current symptoms.
There is no standard treatment for INAD. Diagnosed individuals are managed by a team of medical professionals that recommends treatments based on current symptoms.
After diagnosis, the following evaluations are recommended to determine the extent of disease:
- Ophthalmologic (eye) exam to look for optic atrophy
- EEG to find any unrecognized seizure activity
- Genetics consultation
Supplement
There are good mouse models of INAD and also models of INAD using cells in a test tube. In both the polyunsaturated fatty acid docosahexanoic acid (DHA) has been beneficial. We recommend that people with INAD or PLAN be started on an age or weight appropriate dose of DHA in the form of fish or other oil. Your doctor should recommend the dose and be part of the decision to begin this supplement. DHA is available as a nutritional supplement in most countries.
Long-term management for INAD can include:
- Medication for spasticity, dystonia and/or seizures
- Feeding modifications to prevent aspiration pneumonia and achieve adequate nutrition
- Addition of gastric feeding tube or tracheostomy (if needed)
PROGRESSION
The progression of INAD is usually rapid. Many affected children never learn to walk or lose this ability shortly after learning it. During the last stages of disease, severe spasticity (tight or stiff muscles), progressive cognitive decline and problems with vision have a large impact on daily life.
Many children with INAD do not live beyond age 10, but some do survive into their teens or later ages. Supportive care and symptom management can lead to a longer life span by reducing the risk of infection and other complications.
RESEARCH
You can currently enroll in (or enroll your child in) a natural history study called PLANready. The purpose of this study is to help us better understand the progression of PLAN/INAD and identify disease markers that can be used in future clinical trials. This study can be done completely from home and involves entering information every three-six months. You can learn more about this study and enroll here: PLANREADY
Copyright © 2014 by NBIAcure.org. All rights reserved.