If you or a loved one has recently been diagnosed with BPAN, we hope our website can help answer some of your questions and guide you to the most appropriate resources. We encourage you to call our team to speak to a genetic counselor who can help you navigate this diagnosis. Please email us at info@nbiacure.org and we will set up a phone call with you.
BPAN (Beta-propeller Protein-associated Neurodegeneration) is emerging as the most common NBIA disorder. It is characterized by childhood developmental delay and seizures, with adult-onset movement problems, including dystonia and parkinsonism.
SYMPTOMS
Symptoms of BPAN typically start to appear in early childhood.
Common symptoms include:
Developmental delay
- Usually the first change that is noticed
- Expressive language is significantly affected and kids usually develop few to no words
- Could also be an overall developmental delay
Cognitive (mental) decline
- Can progress to dementia in adulthood
Parkinsonism (symptoms similar to Parkinson’s disease)
- Typically does not start until young adulthood
- Tremors (shaking)
- Bradykinesia (slow movements)
- Rigidity (stiffness)
- Postural instability (loss of balance that causes unsteadiness)
Other muscle problems
- Dystonia (involuntary muscle contraction and spasms)
- Gait freezing (freezing while walking)
- Spasticity (stiff, rigid muscles)
Seizures
- Some children may have multiple seizure types
Abnormal sleep patterns (sleep problems)
Characteristic behaviors and stereotypies (repetitive, rhythmic motions)
- Individuals with BPAN can have some symptoms that are typically associated with Rett syndrome:
- Cognitive (mental) decline with specific loss of expressive language skills
- Hand-wringing
- Seizures
- Abnormal sleep patterns
CAUSE/GENETICS
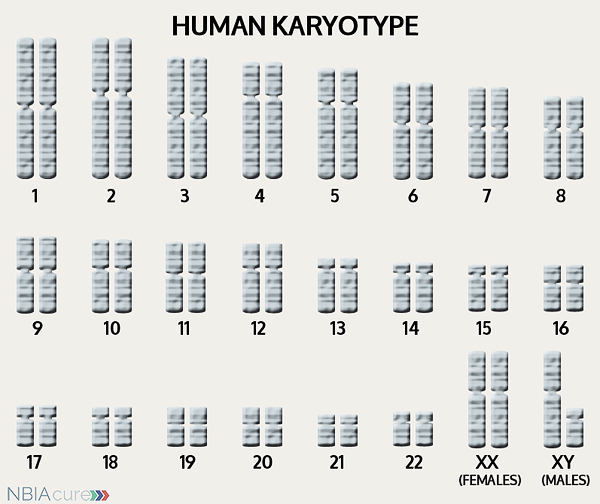 The human body is made up of millions of cells. Inside every cell there is a structure called DNA, which is like an instruction book. DNA contains detailed steps about how all the parts of the body are put together and how they work. However, DNA contains too much information to fit into a single “book”so it is packaged into multiple volumes called chromosomes. Humans typically have 46 total chromosomes that are organized in 23 pairs. There are two copies of each chromosome because we receive one set of 23 chromosomes from our biological mother and the other set of 23 from our biological father. Chromosomes 1-22 are called autosomes and the last pair is called the sex chromosomes because they determine a person’s gender. Females have two X chromosomes and males have one X and one Y.
The human body is made up of millions of cells. Inside every cell there is a structure called DNA, which is like an instruction book. DNA contains detailed steps about how all the parts of the body are put together and how they work. However, DNA contains too much information to fit into a single “book”so it is packaged into multiple volumes called chromosomes. Humans typically have 46 total chromosomes that are organized in 23 pairs. There are two copies of each chromosome because we receive one set of 23 chromosomes from our biological mother and the other set of 23 from our biological father. Chromosomes 1-22 are called autosomes and the last pair is called the sex chromosomes because they determine a person’s gender. Females have two X chromosomes and males have one X and one Y.
Inside every cell there is a structure called DNA, which is like an instruction book. DNA contains detailed steps about how all the parts of the body are put together and how they work. However, DNA contains too much information to fit into a single “book”so it is packaged into multiple volumes called chromosomes. Humans typically have 46 total chromosomes that are organized in 23 pairs. There are two copies of each chromosome because we receive one set of 23 chromosomes from our biological mother and the other set of 23 from our biological father. Chromosomes 1-22 are called autosomes and the last pair is called the sex chromosomes because they determine a person’s gender. Females have two X chromosomes and males have one X and one Y.
If DNA is the body’s instruction book and it is stored in multiple volumes (called chromosomes), then genes would be the individual chapters of those books. Genes are small pieces of DNA that regulate certain parts or functions of the body. Sometimes multiple genes (or chapters) are needed to control one function. Other times, just one gene (or chapter) can influence multiple functions. Since there are two copies of each chromosome, there are also two copies of each gene. In some gene pairs, both copies need to be expressed (or turned on) in order for them to do their job correctly. For other genes pairs, only one copy needs to be expressed.
When a single cell in the human body divides and replicates, its DNA is also replicated. This replication process is usually very accurate but sometimes the body can make a mistake and create a “typo” (or mutation). Just like a typo in a book, a mutation in the DNA can be unnoticeable, harmless, or serious. A mutation with serious consequences can result in a part of the body not developing correctly or a particular function not working properly.
In the case of NBIA disorders, changes in certain genes cause a person to develop their particular type of NBIA. Changes in these NBIA genes lead to the groups of symptoms we observe, although we do not yet understand how the changed genes cause many of these findings. WDR45 is the only gene known to cause BPAN. WDR45’s main job is to tell the body’s cells how to make a protein called WIPI-4, which is involved in the process of autophagy (cells breaking down their own components in order to recycle the parts). It is not yet clear to us how a decrease in this protein eventually leads to iron accumulation in the brain
INHERITANCE
Although BPAN is a genetic condition and the WDR45 gene appears to work in an X-linked dominant pattern, it is usually not inherited from a parent. To understand inheritance and the variability seen in individuals with BPAN, it helps to first understand X-inactivation and mosaicism.
X-inactivation
The WDR45 gene is located on the X chromosome. Females have two X chromosomes, and therefore two copies of the WDR45 gene. Males, in contrast, have one X and one Y chromosome, and only one copy of the WDR45 gene. Since females have an “extra” X chromosome compared to males, their cells only use one copy and the other is “turned off.” This is called X-inactivation. Males have only one X, so they do not need X-inactivation, and they use their single X chromosome in each cell.
This means that a female with BPAN would have some cells in which the WDR45 gene with the mutation is “turned off” and other cells in which the working copy of the gene is “turned off.” This process happens at random and varies from cell to cell. Some females with BPAN may have milder symptoms because they have more cells where the WDR45 mutation has been “turned off” (inactivated). In contrast, males only have one X chromosome. If their single copy of the WDR45 gene has a mutation, then it will always be activated and they will display BPAN symptoms. This is why most males with BPAN likely miscarry during early pregnancy or have more severe symptoms than the females.
Mosaicism
In some rare cases, males and females with BPAN have a relatively high level of function, or mild disease. These individuals may have two types of cells in their bodies: some with a WDR45 mutation and some without. This happens when a normal sperm and egg come together at conception but then as the cells begin to divide, a mutation occurs in the WDR45 gene. Having two cell populations like this is called “mosaicism.” The ratio of cells with or without the mutation depends on at which stage of development and cell division the mutation occurred.
Parents of children with BPAN can also have mosaicism without having BPAN symptoms themselves. Some parents can have mosaicism throughout their bodies, which can be shown by testing blood or skin cells. Others have a very specific type called gonadal mosaicism, where we believe only some of the sperm or egg cells have a genetic change, but we cannot find it elsewhere in the body. We become suspicious for mosaicism or gonadal mosaicism when a healthy couple has more than one child with BPAN, which has been reported multiple times in the literature. Now that we have reviewed the sex chromosomes, X-inactivation, and the concept of mosaicism, it is easier to understand how a child may be born with BPAN:
- Most commonly, it is a new change ONLY in the child with BPAN. The WDR45 gene may have been changed in the sperm or the egg, or it may have happened during or shortly after conception. Typically, they are the only person in the family with BPAN. It is important to test both parents to be as sure as possible that the gene mutation is new.
- Rarely, a couple has more than one child with BPAN. When this occurs, we must assume that the change in WDR45 was inherited from either the mother or father. Either parent could have gonadal mosaicism or mosaicism in additional tissues. It is even possible that the mother could have a WDR45 mutation in ALL her cells, but X-inactivation has turned that copy of the gene off, and it has not affected her health. Sometimes we can prove by testing parental blood samples that one parent has mosaicism or a copy of the WDR45 gene, which helps us understand the chance to have another child with BPAN.
Future Pregnancies
Finally, because the genetics of BPAN is complicated, there are recommendations to consider after having an affected child:
- Both parents of a child with BPAN should always be tested. In the vast majority of cases, this testing will be negative. Although the testing cannot detect all cases of mosaicism, a negative result is reassuring. If a mutation is found in one of the parents, it will give them important information about themselves and possibly other family members, including their other children.
- If a couple has a child with BPAN and additional healthy daughters, their healthy daughters should be counseled as adults and possibly even tested for the WDR45 gene change before they have children of their own. This is a cautious approach that addresses the possibility of a healthy sister inheriting a WDR45 mutation that she does not show due to X-inactivation, but that could be passed on to her children.
- Genetic counseling can help assess the risks in various families and explain complicated concepts like X-inactivation and mosaicism. Prenatal testing is also available during future pregnancies for those who want additional reassurance.
DIAGNOSIS & TESTING
A brain MRI is a standard diagnostic tool for all NBIA disorders. MRI stands for magnetic resonance imaging. An MRI produces a picture of the body that is created using a magnetic field and a computer. The technology used in an MRI is different from that of an x-ray. An MRI is painless and is even considered safe to do during pregnancy. Sometimes an MRI is done of the whole body, but more often, a doctor will order an MRI of one particular part of the body.
Typically, the first indication of a BPAN diagnosis is evidence of brain iron accumulation on a brain MRI. Both T1 and T2 MRIs are necessary for the diagnosis of BPAN.
MRI findings for BPAN include:
- Hypointensity (darkness) in the substantia nigra and globus pallidus on T2 MRI
- The dark patches in the substantia nigra and globus pallidus indicate iron accumulation
- Iron accumulates earlier and to a higher degree in the substantia nigra
- The substantia nigra and cerebral peduncles have a thin, dark central band surrounded by a “halo” of brightness
- Generalized cerebral atrophy (decrease in brain size)
- Mild cerebellar atrophy (decrease in cerebellum size)
- Thinned cerebral peduncle (structure that connects the hindbrain to the forebrain)
Diagnosis of BPAN is confirmed through genetic testing of the WDR45 gene to find a gene change. Genetic testing begins with sequence analysis, and if no gene changes are found, then it continues on to deletion/duplication analysis.
Rarely, an individual with the signs and symptoms of BPAN may not have any WDR45 gene change identified. This can happen because the genetic testing is not perfect and has certain limitations. It does not mean the person does not have BPAN; it may just mean we do not yet have the technology to find the hidden gene change. In these cases it becomes very important to have doctors experienced with BPAN review the MRI and the person’s symptoms very carefully to be as sure as possible of the diagnosis.
MANAGEMENT
 There is no standard treatment for BPAN. Patients are managed by a team of medical professionals that recommends treatments based on current symptoms.
There is no standard treatment for BPAN. Patients are managed by a team of medical professionals that recommends treatments based on current symptoms.
After diagnosis, individuals with BPAN are recommended to get the following evaluations to determine the extent of their disease:
- Neurologic examination for dystonia, rigidity, and spasticity, and parkinsonism
- Evaluation of ambulation and speech
- Developmental assessment
- Assessment for physical therapy, occupational therapy, and/or speech therapy
- Medical genetics consultation
Dystonia (involuntarily muscle contraction and spasms) can be debilitating and distressing to affected individuals and their caregivers. The therapies for managing dystonia vary in method and success rate.
Therapies to manage dystonia can include:
- Intramuscular botulinum toxin
- Botox is injected in spastic, dystonic muscles to help them relax for a period of time
- One of the main drugs used to treat dystonia, usually first taken orally and divided into several doses each day
- In the intrathecal method, an implanted baclofen pump delivers medication directly into the spinal fluid
- Used more often and has some evidence for benefit
- It involves surgical implantation of a lead, extension and battery pack (IPG)
- The lead contains 4 electrodes and is implanted in the globus pallidus region of the brain
- The extension connects the lead to the battery pack (IPG)
- The IPG is a battery-powered neurostimulator that is placed in the abdomen (or in some cases below the clavicle)
- May or may not be indicated for those who are only mildly symptomatic
Medication to manage parkinsonism:
The symptoms of parkinsonism can be treated with the same medications used in Parkinson’s disease. Treatment with dopamine agonist drugs (like Levodopa) must be started and monitored carefully. In the beginning, the dose is increased gradually until both the patient and doctor feel symptoms are under control. While taking dopaminergic drugs, individuals must be regularly monitored for adverse neuropsychiatric effects, psychiatric symptoms and worsening of parkinsonism. There is often short-term great benefit from Parkinsons medications. However, this usually only lasts a few years and is often eventually limited by the development of dyskinesias (a common side effect that creates unwanted movement).
Even after a diagnosis has been made and the appropriate therapies have been chosen, it is recommended to continue long-term surveillance to decrease the impact of BPAN symptoms and increase quality of life.
Long-term surveillance for BPAN can include:
- Medication for spasticity, dystonia, and/or parkinsonism
- Monitoring of individuals receiving dopaminergic drugs for parkinsonism for:
- Adverse neuropsychiatric effects
- Psychiatric symptoms
- Worsening of parkinsonism
- Monitoring of height and weight in children
- Swallowing evaluation and regular dietary assessments
- Assure adequate nutrition
- Prevent aspiration
PROGRESSION
In most individuals, developmental delay and intellectual disability first appear in childhood. Once they reach adolescence or early adulthood, patients often start to regress and cannot regain the skills that they have lost.
The average lifespan varies for individuals with BPAN, but due to improvements in medical care, more affected individuals are now living well into middle age.
RESEARCH
You can currently enroll in (or enroll your child in) a natural history study called BPANready. The purpose of this study is to help us better understand the progression of BPAN and identify disease markers that can be used in future clinical trials. This study can be done completely from home and involves entering information every six months and doing a blood draw once a year. You can learn more about this study and enroll here: BPANREADY
The NBIAcure team hosted a BPAN Research Meeting in Portland from June 24th-June 26th, 2016. To see pictures and review presentations from the symposium, please go here: 2016 BPAN RESEARCH MEETING SUMMARY
Copyright © 2014 by NBIAcure.org. All rights reserved.